Appendix B. Solubility of carbon
dioxide in seawater
Carbon dioxide does not just dissolve in water, rather, it reacts with water to form carbonic acid:
1. CO2 + H2O ↔ H2CO3
The double arrow indicates that the CO2 dissolution reaction is reversible; it can go forward to carbonic acid or backwards to CO2 and water. In time, reversible reactions will reach equilibrium, a point where the forward and backward rates are exactly equal and no net change occurs. Acid-base reactions like those discussed here are very fast and are essentially always at equilibrium. For reactions at equilibrium the concentrations of products and reactants are related by a simple relation:
2. [H2CO3] /([CO2] [H2O]) = constant
The brackets refer to concentration of the species within them. The concentration of water remains essentially unchanged by the dissolution of small amounts of solutes and it is customary to incorporate this constant value into the constant to obtain
3. [H2CO3] / [CO2] = another constant
It is customary to present the concentration of a gas in terms of its partial pressure (PCO2).
4. [H2CO3] / [PCO2] = KH → [H2CO3] = KH·PCO2 = KH·(ppm CO2)/1000000
The constant KH is called the Henry’s Law constant. The Henry’s Law relation shows that carbonic acid concentration is directly proportional to atmospheric CO2 partial pressure. Equation 4 can be expressed in terms of the atmospheric concentration of CO2 in ppm rather than partial pressure. Because atmospheric pressure is one atmosphere (by definition) the partial pressure of a certain ppm level of CO2 is simply that level divided by one million and multiplied by one (atmosphere) as shown in equation 4.
Once in solution carbonic acid dissociates into hydrogen (H+) and bicarbonate (HCO3−) ions:
5. H2CO3 ↔ H+ + HCO3−
Dissociating into hydrogen ions is what makes carbonic acid an acid. Acidity is usually measured by pH which is defined as the negative common logarithm of the hydrogen ion concentration or pH = −log[H+]. Bicarbonate ion is itself an acid and can also dissociate into hydrogen and carbonate (CO32−) ions:
6. HCO3− ↔ H+ + CO32−
Equilibrium relations can be written for reactions 5 and 6 as follows:
7. [H+] [HCO3−] / [H2CO3] = K1
8.
[H+] [CO32-] / [HCO3−] =
Here K1 and
9. [HCO3−] = K1 [H2CO3] / [H+]
10. [CO32-] =
The sum of carbonic acid, bicarbonate and carbonate species is then given by:
11. dissolved
CO2 = [H2CO3] + [HCO3−]
+ [CO32-] = [H2CO3]∙(1 + K1/
[H+] + K1·
= KH·(ppm
CO2)∙(1 + K1/ [H+] + K1·
Equation 11 gives the solubility of CO2 in seawater as a function of [H+] and ppm CO2 in the atmosphere for a given set of constant values. Since the constants themselves are functions of temperature, CO2 solubility also depends on temperature through temperature’s effect on the constants.
The pH of the oceans is about 8.1. Thus [H+] = 10−8.1 = 7.94 x 10-9 moles/liter. This value was put into equation 11 with constants K1 and K2 obtained from Roy1 et al. 1993, and KH obtained from Weiss2 (1974). A value of 0.0022 mol/kg (2.3 millimolar) was obtained for CO2 solubility in seawater at 20º C. This value is close to 2.1-2.3 millimolar value given in the literature.3-4 For my purposes, I am interested in how CO2 solubility changed with rising CO2 levels over the last 50 years, the exact values themselves are not so critical, but rather how rising CO2 level has affected solubility and hence the ability of the oceans to absorb CO2. To use equation 11 to answer this question it is necessary to determine how changes in dissolved CO2 affect [H+] (pH). Once this is known then calculation of solubility is straightforward.
An initial attempt to deal with this issue is to consider the case of CO2 dissolution in pure water to form H2CO3. Some of the H2CO3, will disassociate into H+ and HCO3− according to equation 5. The amount of each will be equal, and we can represent the concentration of each as x. With this equation 7 can be written as follows
12. x2 / [H2CO3] = K1 → x = (K1 [H2CO3] )½ = [H+]
[H2CO3] can be calculated from ppm CO2 using equation 4 and the result substituted into equation 12 to give:
13. [H+] = (K1∙∙KH∙ppm CO2)½/1000
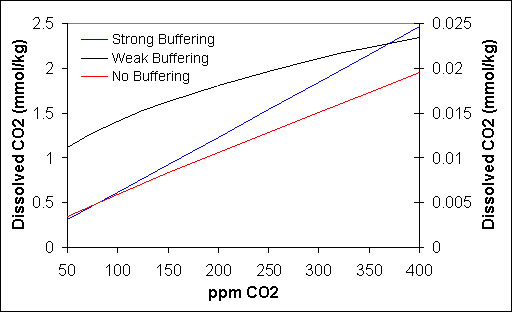
Equation 13 can be used to calculate [H+] which is then used in equation 11 to determine CO2 solubility. This was done and the results appear as the red line in Figure 1. The solubility of CO2 in pure water is very low, more than 100 times less than the solubility in seawater. This is because in pure water, the acidity of carbonic acid is sufficiently strong to drive the pH well into the acid range. Using constants for pure water and 25º C, equation 13 gives pH 5.6 for the present-day CO2 level of 380 ppm, well below the ocean pH of 8.1. Indeed the pH of rainwater in clean air is about 5.6, reflecting dissolution of atmospheric carbon dioxide. Polluted air containing oxides of sulfur or nitrogen, which upon dissolution form acids much stronger than carbonic acid, creates the phenomenon known as acid rain, in which the pH of rainwater can fall to 3 or even lower.
Acid rain occurs because pure water has no buffering. That is, it contains no alkaline substances that react with dissolved acids to counteract their acidity. The ocean contains such buffering, which is why its pH is high (8.1) in the presence of dissolved carbon dioxide. Obviously the simple case of CO2 dissolution in water is not a useful model for determining the effect of CO2 levels on seawater pH. Another simple approach would be to assume that the buffering is strong enough to keep the pH from changing because of the dissolution of a weak acid like CO2. In this case CO2 solubility will be directly proportional to ppm CO2 in the atmosphere as shown by the blue line in Figure 1. In actuality, pH has fallen about 0.075 unit since the beginning of the industrial revolution largely because of increasing atmospheric CO2.5-7 Apparently, the ocean is not strongly buffered. Looking at the composition of seawater, two buffering agents are present.3 These agents must govern the pH of seawater. They are calcium carbonate (CaCO3) and the dihydrogen borate ion (H2BO3−) which react with carbonic acid according to the following reactions:
14. H2CO3 + CaCO3 ↔ Ca2+ + 2 HCO3−
15. H2CO3 + H2BO3− ↔ H3BO3 + HCO3−
Equilibrium expressions can be written for these reactions as before:
16. [Ca2+]∙[HCO3−]2
/ [H2CO3] = KC
= K1 KSP /
17. [HCO3−]∙[H3BO3] / ([H2BO3−]∙[H2CO3]) = KB = K1 / BK1
The equilibrium constants are
related to the carbonic acid dissociation constants K1 and
I desire to calculate [H+] as CO2 levels change from the standard level of 370 ppm. Equation 4 gives [H2CO3] directly. Given a value for [HCO3−] equation 9 can be solved for [H+]. Thus the problem becomes determining how the simultaneous equilibria expressed in equation 16 and 17 affect [HCO3−] as [H2CO3] changes. This value can be expressed in terms of deviations from the standard values caused by the buffering reactions 14 and 15. If I denote x as the extent to which reaction 14 moves forward and y as the extent to which reaction 15 moves forward, I can write equations 16 and 17 as:
18. ([Ca2+]0 + x)∙([HCO3−]0 + 2x + y)2 / [H2CO3] = KC
19. ([HCO3−]0 + 2x + y)∙([H3BO3]0 + y) / {([H2BO3−]0 − y)∙[H2CO3]} = KB
Here the subscript 0 refers to the base values for the various concentrations, which were previously calculated for 370 ppm CO2 and pH 8.1. No subscript appears for [H2CO3] because this concentration is determined from the atmospheric CO2 level (which is a given) according to equation 4. Any H2CO3 consumed or produced by the reactions 14 and 15 would quickly be made up by CO2 exchange with the atmosphere according to reaction 1. With this, all parameters in equations 18 and 19 except x and y are known and these equations can be solved for x and y. This was done by first solving equation 18 algebraically for y in terms of x. With this, for a given value of x, a value of y was determined and this value, plus the assumed x value was used in equation 19. Using the Microsoft Excel goal seek function, the value x need to solve equation 19 can be found readily. With x and y known, so is [HCO3−], from which [H+] is obtained from equation 7. With [H+], CO2 solubility is calculated according to equation 11. The result appears in Figure 1 as the weak buffering line.
As a test of the validity of this model, pH values were calculated for the CO2 level in the year 1751 (277 ppm) and for 1994 (359 ppm). A temperature of 14º C was used for the 1751 value (as compared to 15º C for today) to account for the roughly one degree of warming since then. Values of 8.173 and 8.108 were obtained for pH, compared to literature values of 8.179 and 8.104.5-7 This close correspondence is a good indication that the model presented here is valid.
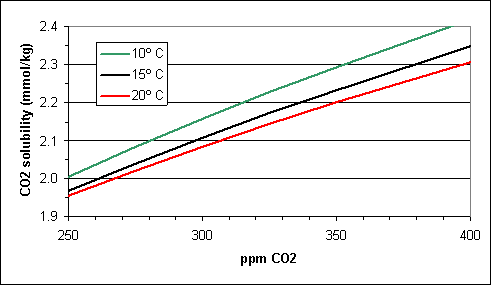
With a valid model it is instructive to use it to examine a number of issues raised in the global warming debate. One of these is the “soda pop analogy”. Some global warming skeptics like to argue that higher CO2 levels today are the result of higher temperatures and not the other way around. They like to use the analogy that, just as warm soda goes flat (loses its dissolved CO2), the oceans have lost some of their vast reserves of dissolved CO2 as a result of the roughly 1º C degree of warming since the beginning of the industrial revolution. Using equations 18 and 19 and the goal seek function, it is a straightforward matter to calculate CO2 solubility as a function of CO2 for various temperatures (see Figure 2). From Figure 2 it is clear that the solubility at 380 pm CO2 (today’s value) at a temperature of 20º C (5º C higher than today) is higher than the solubility at 300 ppm CO2 (the value a century ago) at a temperature of 10º C (more than 4º C lower than then). That is, even if the temperature rise were ten times greater than it actually was, they would still be no net release of CO2 from the oceans over the last century, but rather a net absorption of CO2.
In actual fact, only about half of the CO2 released to the atmosphere by human activity over the past century is still in the atmosphere. If the missing CO2 was removed by the oceans, we can use the solubility model presented here to determine the amount of seawater (i.e. the depth of the surface layer of the ocean) necessary to absorb the missing CO2.
References
1.
Roy, R.N.L., N. Roy, M. Vogel, C. P. Moore, T.
Pearson, C. E. Good, F. J. Millero, and D. J.
Campbell, (1993) “Determination of the ionization constants of carbonic acid in
seawater”, Marine Chemistry, 44 249-268. (www.ugamp.nerc.ac.uk/um/source_p/umpl/EQ_CONST_dk.htm)
2. Weiss,
R.F. (1974) "Carbon dioxide in water and seawater: the solubility of a
non-ideal gas" Mar. Chem. 2: 203-2 15. (www.ugamp.nerc.ac.uk/um/source_p/umpl/EQ_CONST_dk.htm)
3. Anthoni,
J F., “Composition of seawater”, (www.seafriends.org.nz/oceano/seawater.htm)
4. Classroom Bermuda Atlantic Time Series Study, “How
much CO2 can sea water hold?”, (www.coexploration.org/bbsr/classroombats/html/co2_in_the_sea.html)
5. “Oceanic acidification” Wikipedia (http://en.wikipedia.org/wiki/Ocean_acidification)
6. Orr, J. C. et al. (2005). “Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms” Nature 437, 681-686.
7. Key,
R.M., Kozyr, A., Sabine, C.L., Lee, K., Wanninkhof, R., Bullister, J.,
Feely, R.A., Millero, F., Mordy, C. and Peng, T.-H.
(2004). A global ocean carbon climatology: Results from GLODAP. Global
Biogeochemical Cycles 18, GB4031.
8. Klostermann, A F. “The Calcium Question”, (www.natureef.com/calcium1.htm)
9. Dickson,
A.E. (1990) “Thermodynamics of the dissociation of boric acid in syntheic sea water from 273.15 to 298.15 K”, Deep-Sea Research, 37, 755-766. (www.ugamp.nerc.ac.uk/um/source_p/umpl/EQ_CONST_dk.htm)
10. World
Resources Institute Earthtrends database (http://earthtrends.wri.org/searchable_db/index.php?theme=3)
{kind=link}
{kind=link}